Treatment guidelines (disease severity classification / disease activity index)
I. Castleman Disease
Diagnosis of Castleman Disease
A pathological diagnosis is crucial for making accurate diagnosis of Castleman disease. In principle, the diagnosis requires an evaluation of the lymph node. The lymph node subjected to biopsy should be decided based on results obtained by physical examinations, full-body CT or FDG-PET (not covered by insurance) scans, which determine the size and distribution of the lymph node lesion(s).
Tentative diagnostic criteria proposed by this research policy group1,2)
Essential diagnostic criteria for Castleman disease (both A and B must be met)
A Both of the following conditions should be met:
1. Single or multiple swollen lymph node/s (the longest diameter >1 cm) should be present.
2. Histopathological findings of the affected lymph node(s) or organ(s) should be consistent with one of the following histological types of Castleman disease:
1) Hyaline-vascular type
2) Plasma cell type
3) Mixed type
B The following diseases should be excluded as the cause of lymphadenopathy.
- Malignant neoplasms: Angioimmunoblastic T-cell lymphoma, Hodgkin lymphoma, follicular dendritic cell sarcoma, renal cell carcinoma, malignant mesothelioma, lung cancer, cervical cancer, etc.
- Infectious diseases: Nontuberculous mycobacterial infection, cat scratch disease, rickettsial disease, toxoplasmosis, fungal infection, infectious mononucleosis, chronic active Epstein-Barr (EB) virus infection, acute HIV infection, etc.
- Autoimmune diseases: Systemic lupus erythematosus, Sjögren’s syndrome, etc.
- Other diseases manifesting symptoms similar to Castleman disease: IgG4-related diseases, histiocytic necrotizing lymphadenitis, sarcoidosis, idiopathic portal hypertension, etc.
Reference for diagnosis
- Castleman disease can present with various clinical symptoms of different degrees of severity. Frequent symptoms include low- to moderate-grade fever, general malaise, easy fatigability, weight loss, night sweats, and superficial lymphadenopathy. Some cases manifest with skin lesions (flat or slightly elevated brown or dark red eruptions, pemphigoids, xanthoma, atopic dermatitis, or hemangiomas), abdominal distension, edema, shortness of breath, dyspnea, and hemorrhagic tendency. Castleman disease can be associated with vascular events such as cerebral infarction, and peripheral neuropathy.
- In addition to lymphadenopathy, imaging examinations may reveal hepatosplenomegaly, ascites, pleural effusion, and pulmonary interstitial shadows.
- Blood tests usually show a positive CRP and elevated serum IL-6 levels. Microcytic anemia and thrombocytosis are frequently observed. Decreased serum LDH and albumin levels, and increased serum alkaline phosphatase, IgE, and VEGF levels are also frequently observed. Polyclonal hypergammaglobulinemia is another important feature of Castleman disease. Autoantibodies, such as anti-nuclear antibody, are occasionally detected.
- In some cases, renal dysfunction, interstitial pulmonary lesions, pulmonary hypertension, dilated cardiomyopathy, autoimmune thrombocytopenia, autoimmune hemolytic anemia, endocrinopathy (such as hypothyroidism), and/or AA-amyloidosis accompany the disease.
- Elevation of serum IgG4 levels or increased IgG4 positive cells in the lymph nodes can be observed, not only in IgG4-related disease, but also in Castleman disease. Presence of fever, microcytic anemia, thrombocytosis, and elevations of serum CRP, which are caused by overproduction of IL-6, are signs supporting the diagnosis of Castleman disease rather than IgG4-related disease.
- Human herpesvirus type 8 (human herpesvirus-8, HHV-8; Kaposi's sarcoma-associated herpesvirus, KSHV) associated multicentric Castleman disease can be diagnosed by the typical histopathological features and the presence of the HHV-8 genome in the blood or in the lymph nodes. Majority of patients with HHV-8-associated Castleman disease are HIV positive, and Kaposi’s sarcoma occasionally develops simultaneously or subsequently.
- POEMS syndrome is a disease entity with monoclonal gammopathy accompanied by polyneuropathy, organomegaly, and endocrinopathy. This entity is believed to be a relative of multiple myeloma, but a part of its pathological conditions overlaps with those of Castleman disease. POEMS syndrome is not excluded from Castleman disease in this classification.
- TAFRO syndrome is a medical condition or disorder presenting with thrombocytopenia, anasarca, fever, bone marrow fibrosis, and organomegaly. Histology of lymph nodes from patients with this syndrome is indistinguishable from that of Castleman disease. In this classification, TAFRO syndrome is not excluded from Castleman disease.
Clinical disease type classification 1,2)
Unicentric Castleman disease, (UCD) where the lesion is confined to one region is distinguished from multicentric Castleman disease (MCD) spreading over multiple regions. MCD is further classified into HHV-8-associated MCD caused by infection with HHV-8 and MCD without HHV-8 infection, which is referred to as idiopathic MCD (idiopathic MCD, iMCD) (Figure below).
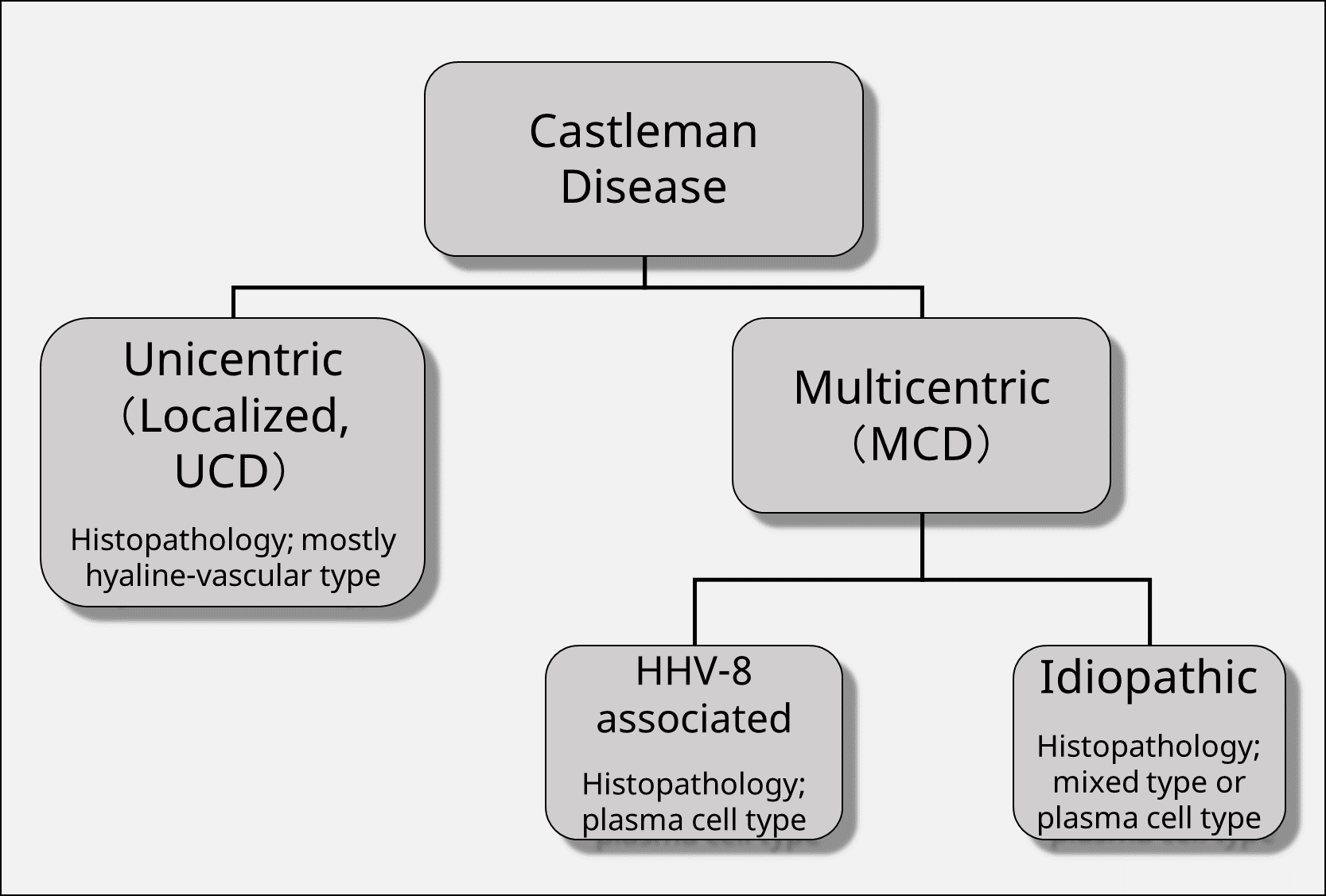
Disease classification by Castleman Disease Collaborative Network (CDCN) and diagnostic criteria for iMCD
In CDCN, POEMS syndrome is independently illustrated and, based on pathological tissue classification, classification of Castleman disease is proposed as shown in the figure below 3).
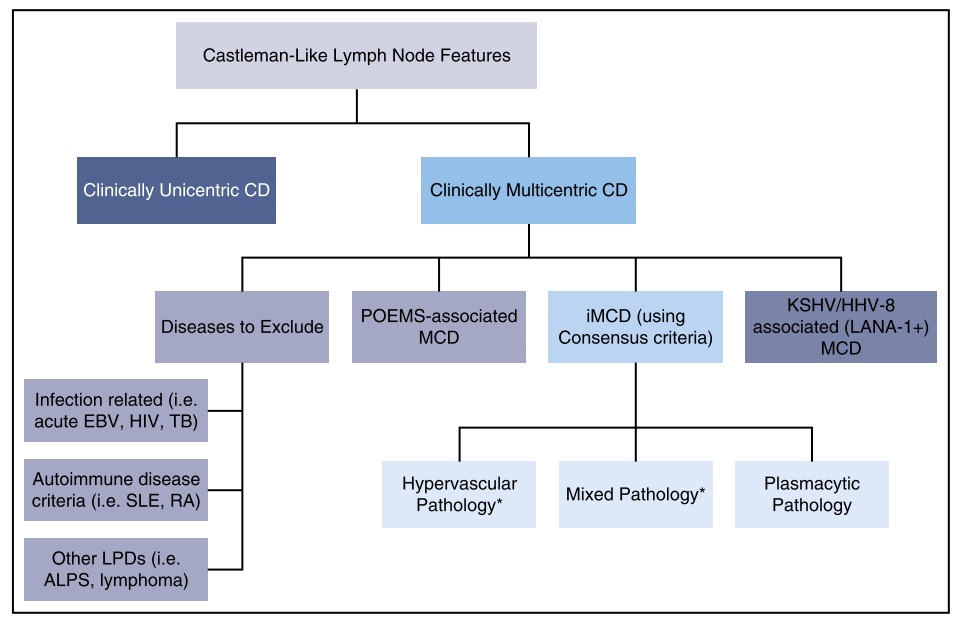
Adapted from Figure4 of Ref. 3.
With respect to iMCD, the following international diagnostic criteria have been proposed3).
International diagnostic criteria for iMCD by CDCN 3
iMCD is diagnosed when satisfying two major criteria and two or more items including at least one of the test criteria out of the 11 small criteria as well as diseases defined by exclusion criteria are excluded.
Consensus diagnostic criteria for iMCD (*Adapted from ref. 3 table 2)
I. Major Criteria (need both):
- Histopathologic lymph node features consistent with the iMCD spectrum. Features along the iMCD spectrum include (need grade 2-3 for either regressive GCs or plasmacytosis at minimum):
・Regressed/atrophic/atretic germinal centers, often with expanded mantle zones composed of concentric rings of lymphocytes in an “onion skinning” appearance
・FDC prominence
・Vascularity, often with prominent endothelium in the interfollicluar space and vessels penetrating into the GCs with a “lollipop” appearance
・Sheetlike, polytypic plasmacytosis in the interfollicular space
・Hyperplastic GCs - Enlarged lymph nodes (≥1 cm in short-axis diameter) in ≥2 lymph node stations
II. Minor Criteria (need at least 2 of 11 criteria with at least 1 laboratory criterion) Laboratory*
- Elevated CRP (>10 mg/L) or ESR (>15 mm/h)†
- Anemia (hemoglobin <12.5 g/dL for males, hemoglobin <11.5 g/dL for females)
- Thrombocytopenia (platelet count <150 k/mL) or thrombocytosis (platelet count >400 k/mL)
- Hypoalbuminemia (albumin <3.5 g/dL)
- Renal dysfunction (eGFR <60 mL/min/1.73m2) or proteinuria (total protein 150 mg/24 h or 10 mg/100 ml)
- Polyclonal hypergammaglobulinemia (total γ globulin or immunoglobulin G >1700 mg/dL)
Clinical
- Constitutional symptoms: night sweats, fever (>38°C), weight loss, or fatigue (≥2 CTCAE lymphoma score for B-symptoms)
- Large spleen and/or liver
- Fluid accumulation: edema, anasarca, ascites, or pleural effusion
- Eruptive cherry hemangiomatosis or violaceous papules
- Lymphocytic interstitial pneumonitis
III. Exclusion Criteria (must rule out each of these diseases that can mimic iMCD) Infection-related disorders
- HHV-8 (infection can be documented by blood PCR, diagnosis of HHV-8–associated MCD requires positive LANA-1 staining by IHC, which excludes iMCD)
- Clinical EBV-lymphoproliferative disorders such as infectious mononucleosis or chronic active EBV (detectable EBV viral load not necessarily exclusionary)
- Inflammation and adenopathy caused by other uncontrolled infections (eg, acute or uncontrolled CMV, toxoplasmosis, HIV, active tuberculosis)
Autoimmune/autoinflammatory diseases (requires full clinical criteria, detection of autoimmune antibodies alone is not exclusionary)
- Systemic lupus erythematosus
- Rheumatoid arthritis
- Adult-onset Still disease
- Juvenile idiopathic arthritis
- Autoimmune lymphoproliferative syndrome
Malignant/lymphoproliferative disorders (these disorders must be diagnosed before or at the same time as iMCD to be exclusionary):
- Lymphoma (Hodgkin and non-Hodgkin)
- Multiple myeloma
- Primary lymph node plasmacytoma
- FDC sarcoma
- POEMS syndrome‡
Select additional features supportive of, but not required for diagnosis
Elevated IL-6, sIL-2R, VEGF, IgA, IgE, LDH, and/or B2M
Reticulin fibrosis of bone marrow (particularly in patients with TAFRO syndrome)
Diagnosis of disorders that have been associated with iMCD: paraneoplastic pemphigus, bronchiolitis obliterans organizing pneumonia, autoimmune cytopenias,
polyneuropathy (without diagnosing POEMS‡), glomerular nephropathy, inflammatory myofibroblastic tumor
B2M, β-2-microglobulin; CMV, cytomegalovirus; CTCAE, common terminology for adverse events; eGFR, estimated glomerular filtration rate; GC, germinal center; IHC,
Immunohistochemistry; LANA-1, latency-associated nuclear antigen; LDH, lactate dehydrogenase.
*We have provided laboratory cutoff thresholds as guidance, but we recognize that some laboratories have slightly different ranges. We suggest that you use the upper and lower ranges from your particular laboratory to determine if a patient meets a particular laboratory Minor Criterion.
†Evaluation of CRP is mandatory and tracking CRP levels is highly recommended, but ESR will be accepted if CRP is not available.
‡POEMS is considered to be a disease “associated” with CD. Because the monoclonal plasma cells are believed to drive the cytokine storm, we do not consider it iMCD, but rather “POEMS-associated MCD.”
Severity classification
The research group has proposed a system to classify the severity of Castleman disease into three stages of mild, moderate, and severe depending mainly on organ disorders related to life prognosis. At the meeting of our research group on June 9, 2018, we modified slightly from the severity classification of the article published in Rinsho Ketsueki, “A reference guide for management of Castleman― disease”. The latest severity classification system proposed by our group is shown below. It is a provisional classification and opinions will be highly appreciated.
| Severe | Moderate | Mild | |
|---|---|---|---|
| Anemia of inflammation | Hb <7g/dL or red blood cell transfusion dependent | 7 g/dL ≤Hb <8 g/dL | Not applicable to any on the left. |
| Thrombocytopenia | Platelet transfusion-dependent or -refractory symptomatic thrombocytopenia | Platelet counts <20×103/μL | |
| Hypoalbuminemia | Alb <1.5 g/dL | 1.5 ≤Alb <2.0 g/dL | |
| Renal dysfunction | GFR <15ml/minute/1.73m² or nephrotic syndrome |
Red in CKD severity classification heat map | |
| Pulmonary lesion | Presence of interstitial pulmonary lesions and requires oxygen inhalation at rest | Presence of interstitial pulmonary lesions and difficulty breathing with daily light exertion. | |
| Anasarca | Massive pleural effusion or ascites requiring occasional drainage to relieve symptoms | Pleural effusion or ascites shown by imaging tests | |
| Heart Failure | EF <40% or NYHA IV degrees | 40% ≤EF <50% orNYHA III degree | |
| Amyloidosis | Organ damages due to histologically proven secondary amyloidosis |
CKD severity classification heat map
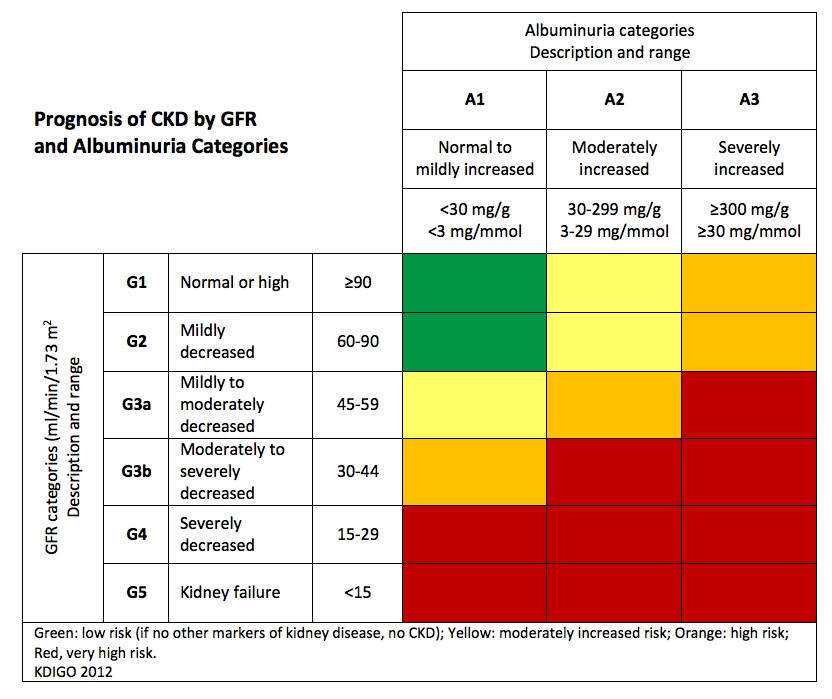
Adapted from website of National Kidney Foundation (https://www.kidney.org/kidneydisease/siemens_hcp_quickreference)
Disease activity evaluation system
In our research group, we propose a CHAP score as an indicator of disease activity 2). This is the sum of CRP, hemoglobin (Hb), serum albumin, performance status (PS) scores, and disease activity is evaluated from 0 point up to 16 points. It is assumed that the CHAP score will be used for evaluating change of disease conditions as well as treatment effects in the same patient.
| Score | 0 | 1 | 2 | 3 | 4 |
|---|---|---|---|---|---|
| CRP (mg/dL) |
<1 | ≥1, <5 | ≥5, <10 | ≥10, <20 | ≥20 |
| Hb (g/dl) |
≥12 | <12, ≥10 | <10, ≥8 | <8 | Transfusion dependent |
| Albumin (g/dL) |
≥3 | <3, ≥2.5 | <2.5, ≥2 | <2, ≥1.5 | <1.5 |
| P. S. (ECOG) |
0 | 1 | 2 | 3 | 4 |
Treatment for CD
(1)Unicentric (localized) Castleman disease(UCD)
In most cases, cure can be expected by local therapy (such as surgical resection). It is important to completely remove the lesion. If a recurrence occurs after surgical resection, or if it is difficult to resect the entire lesion and systemic inflammatory symptoms are present, treatment strategies for idiopathic MCD may be conducted.
(2)Idiopathic multicentric Castleman disease (iMCD)
Patients with no or modest symptoms may be observed without treatments; but in many cases, therapeutic intervention is necessary to alleviate symptoms such as fatigue 4). When the symptoms of systemic inflammation are mild, the symptoms may be alleviated with low to intermediate doses of corticosteroids (about 0.3 mg/kg prednisolone in the absence of signs of organ damage, about 0.5 to 1 mg/kg when organ damages are observed). After the symptoms are improved, the dose of corticosteroids should be gradually tapered. In the cases with prolonged administration of corticosteroids, close attention should be paid to the onset of diabetes, osteoporosis, and infections caused by herpes virus and fungi.
Treatment with tocilizumab (an anti-interleukin 6 receptor antibody) should be considered in cases with severe inflammatory symptoms or organ damage in the kidney, the lung and the others (moderate to severe disease in the severity classification system). Even cases with mild disease, administration of tocilizumab may be considered when the progression of clinical symptoms or organ disorders cannot be sufficiently controlled with steroid alone. Before starting administration of tocilizumab, physicians should explain the possibility of lifelong needs of its administration to patients. Usually, 8 mg/kg of tocilizumab is administered intravenously every two weeks (the dosing interval can be shortened up to one week depending on symptoms). Tocilizumab may be used as an initial treatment in combination with corticosteroids, or alone when corticosteroid treatment is inappropriate due to comorbidities or other reasons. In many cases, once treatment with tocilizumab is started, various systemic inflammatory symptoms and abnormal laboratory values promptly improve. It is often possible to reduce/discontinue the dose of steroids that are used in combination. Also, the enlarged spleen and lymph nodes may gradually shrink. Side effects include headache, upper respiratory tract inflammation, itching, rash, and allergies. Many of these are minor, but serious infections such as pneumonia and septicemia have also been reported5,6). During the treatment with tocilizumab, CRP levels will not increase even in serious infection; therefore, care must be taken not to overlook infectious diseases. Anaphylaxis has been observed in over 1% of cases. Generally, treatment with tocilizumab should not be discontinued once started, but if it is inevitable to halt, temporarily administer or increase steroid to prevent the rebound of inflammatory symptoms. When the treatment with tocilizumab is started, the interleukin 6 concentration in the serum (insurance-inapplicable examination in Japan) jumps up because the interleukin 6 cannot bind to the receptor and is not removed from the plasma6).
Siltuximab, an antibody preparation against interleukin 6, has a therapeutic effect comparable to that of tocilizumab and was approved by the US FDA in 2014 (not approved in Japan). It is currently the only drug validated in a randomized controlled trial7).
In cases where it is refractory or intolerant to treatment with corticosteroids or tocilizumab, and it is difficult to control the disease state, steroid pulse therapy and chemotherapies for malignant lymphoma and multiple myeloma have been attempted (not covered by insurance)8-10).
(3)HHV-8 associated MCD
It is a subtype found primarily in HIV-infected patients, but it is occasionally seen in HIV-negative patients with immunodeficiency11). Currently, this subtype is considered extremely rare in Japan12,13). This disease type develops rapidly with inflammatory symptoms of the whole body and often complicates Kaposi's sarcoma or malignant lymphoma11). Although survival exceeding 2 years was rare, recently, very promising results with combination therapies with high dose zidovudine (azidothymidine, AZT) + valganciclovir + rituximab or rituximab + pegylated liposomal doxorubicin + anti-HIV drug have been reported14,15). Interferon or high dose AZT is used as maintenance therapy after remission.
References
- Yoshizaki K, Okamoto S, Kawabata H, et al: A reference guide for management of Castleman disease. Rinsho Ketsueki 58: 97-107, 2017
- Fujimoto S, Koga T, Kawakami A, et al: Tentative diagnostic criteria and disease severity classification for Castleman disease: A report of the research group on Castleman disease in Japan. Mod Rheumatol 28: 161-167, 2018
- Fajgenbaum DC, Uldrick TS, Bagg A, et al: International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 129: 1646-1657, 2017
- Liu AY, Nabel CS, Finkelman BS, et al: Idiopathic multicentric Castleman's disease: a systematic literature review. The Lancet Haematology 3: e163-175, 2016
- Nishimoto N, Terao K, Mima T, et al: Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 112: 3959-3964, 2008
- Chugai Pharmaceutical Co., Ltd: アクテムラ全例調査の中間報告(キャッスルマン病)[An interim report of the post-marketing surveillance of ActemraTM for Castleman disease], 2014, http://chugai-pharm.jp/hc/ss/pr/drug/act_via0400/report/scs/PDF/20140225_act_epmpv_safe_cd.pdf,
- van Rhee F, Wong RS, Munshi N, et al: Siltuximab for multicentric Castleman's disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol 15: 966-974, 2014
- Soumerai JD, Sohani AR, Abramson JS: Diagnosis and management of Castleman disease. Cancer Control 21: 266-278, 2014
- Seo S, Yoo C, Yoon DH, et al: Clinical features and outcomes in patients with human immunodeficiency virus-negative, multicentric Castleman's disease: a single medical center experience. Blood Res 49: 253-258, 2014
- Cervantes CE, Correa R: Castleman Disease: A Rare Condition with Endocrine Manifestations. Cureus 7: e380, 2015
- Parravicini C, Corbellino M, Paulli M, et al: Expression of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative Castleman's disease. Am J Pathol 151: 1517-1522, 1997
- Kojima M, Nakamura N, Tsukamoto N, et al: Clinical implications of idiopathic multicentric castleman disease among Japanese: a report of 28 cases. Int J Surg Pathol 16: 391-398, 2008
- Kawabata H, Kadowaki N, Nishikori M, et al: Clinical Features and Treatment of Multicentric Castleman's Disease : A Retrospective Study of 21 Japanese Patients at a Single Institute. J Clin Exp Hematop 53: 69-77, 2013
- Uldrick TS, Polizzotto MN, Yarchoan R: Recent advances in Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Curr Opin Oncol 24: 495-505, 2012
- Uldrick TS, Polizzotto MN, Aleman K, et al: Rituximab plus liposomal doxorubicin in HIV-infected patients with KSHV-associated multicentric Castleman disease. Blood 124: 3544-3552, 2014
II TAFRO Syndrome
1)Diagnostic criteria for TAFRO Syndrome
Research team for establishment of TAFRO syndrome, which was funded by the 2015 fiscal year Grant-in-Aid for Scientific Research Grant (Intractable Disease Policy Research Project) New diseases, has published the 2015 version of TAFRO syndrome diagnostic criteria as follows.
TAFRO Syndrome Diagnostic Criteria 2015
* The 2015 diagnostic criteria for TAFRO syndrome, as determined by All Japan TAFRO Syndrome Research Group in the Research Program for Intractable Disease by Ministry of Health, Labor and Welfare (MHLW) Japan
[Disease description]
TAFRO syndrome is a systemic inflammatory disorder manifesting as fever; anasarca including pleural effusion and ascites; thrombocytopenia; renal insufficiency; anemia; and organomegaly including hepatosplenomegaly and lymphadenopathy. It has an acute or sub-acute onset of unknown etiology. The syndrome first reported by Takai, et al. in 2010, includes Thrombocytopenia, Anasarca, Fever, Reticulin fibrosis, and Organomegaly; other patients with similar symptoms have been reported. Although several clinical and pathological findings of TAFRO syndrome resemble those of Castleman disease, presence of other specific features that differentiate TAFRO syndrome and Castleman disease are now being discussed. Several patients have been successfully treated with glucocorticoids, immunosuppressants including cyclosporin A, tocilizumab or rituximab, whereas others were refractory to treatment and took a fatal clinical course. Reliable early diagnosis and appropriate rapid treatment are therefore essential for favorable outcomes.
【Diagnostic criteria of TAFRO syndrome】
・A diagnosis of TAFRO syndrome requires all of the three major categories and at least two of four minor categories.
・As it is very important to exclude malignancies, including lymphoma, lymph node biopsy, if applicable, is strongly recommended.
A. Major categories
1) Anasarca, including pleural effusion, ascites and general edema
2) Thrombocytopenia; defined as a pre-treatment platelet count ≤100,000/μl.
3) Systemic inflammation, defined as fever of unknown etiology above 37.5℃ and/or serum C-reactive protein concentration ≥2mg/dl.
B. Minor categories
1) Castleman disease-like features on lymph node biopsy
2) Reticulin myelofibrosis and/or increased number of megakaryocytes in bone marrow
3) Mild organomegaly, including hepatomegaly, splenomegaly and lymphadenopathy 4) Progressive renal insufficiency
C. Diseases to be excluded
1) Malignancies, including lymphoma, myeloma, mesothelioma, et ceteta.
2) Autoimmune disorders, including systemic lupus erythematosus (SLE), ANCA-associated vasculitis, et ceteta.
3) Infectious disorders, including acid fast bacterial infection, rickettsial disease, lyme disease, severe fever with thrombocytopenia syndrome (SFTS), et cetera.
4) POEMS syndrome
5) IgG4-related disease
6) Hepatic cirrhosis
7) Thrombotic thrombocytopenic purpura (TTP)/ hemolytic uremic syndrome (HUS)
D. Points to consider
・Marked polyclonal hypergammopathy is rare in TAFRO patients, with serum IgG concentrations remaining below 3,000 mg/dl.
・Obvious monoclonal protein should not be present.
・Few patients show elevated serum LDH.
・Most patients show elevated level of serum ALP.
・Hepatosplenomegaly in this disease is usually mild and only confirmed by CT-scan, whereas presence of huge hepatosplenomegaly may indicate lymphoma and other diseases.
・Lymphadenopathy in this disease is usually smaller than 1.5 cm in diameter, whereas huge lymphadenopathy may indicate lymphoma and other diseases.
・Exclusion criteria for Castleman disease and immune thrombocytopenia (ITP) have not been determined, so these diseases may not be excluded at present.
3)Severity classification of TAFRO syndrome and disease activity index
Severity classification has also been published from the same research group, and it can be used to evaluate treatment effectiveness and disease activity.
[The 2015 disease severity classification for TAFRO syndrome]
Total points are calculated by adding the points of present symptoms.
1) Anasarca: three points maximum
One point for pleural effusion on imaging
One point for ascites on imaging
One point for pitting edema on physical examination
2) Thrombocytopenia: three points maximum
One point for lowest platelet counts < 100,000/μl
Two points for lowest platelet counts < 50,000/μl
Three points for lowest platelet counts < 10,000/μl
3) Fever and/or inflammation: three points maximum
One point for fever ≥37.5℃ but < 38.0℃ or for CRP ≥ 2mg/dl but < 10mg/dl
Two points for fever ≥38.0℃ but < 39.0℃ or for CRP ≥ 10mg/dl but < 20mg/dl
Three points for fever ≥39.0℃ or for CRP ≥ 20mg/dl
4) Renal insufficiency: three points maximum
One point for GFR< 60ml/min/1.73m2
Two point for GFR< 30ml/min/1.73m2
Three points for GFR< 15ml/min/1.73m2 or need for hemodialysis
Relationship between score and disease severity
0-4 points: mild (grade 1)
5-6 points: moderate (grade 2)
7-8 points: slightly severe (grade 3)
9-10 points: severe (grade 4)
11-12 points: very severe (grade 5)
4.)Treatment guidelines for TAFRO syndrome
The 2015 treatment strategy for TAFRO syndrome
- High-dose glucocorticoid: prednisolone 1mg/kg/day for two weeks, followed by tapering; OR Methyl-prednisolone pulse therapy with 500-1000mg/day for three days if an emergency.
- CyclosporinA (CsA) : May be added for patients refractory or dependent on glucocorticoids. The starting dose of oral CsA is 3 to 5mg/kg/day, divided into two doses per day. The target trough level (C0) of serum CsA is 150- 250 ng/ml. However, measurement of serum concentration at 2 hours after taking medicine (C2) is recommended. Patients with C2 level of CsA < 600 ng/ml, suggesting an insufficient serum concentration, should be switched from after-meal to before-meal administration. If serum creatinine level increase > 150%, CsA dose should be decreased 50% to 75%.
- Tocilizumab (anti-IL-6 receptor antibody): for patients with TAFRO syndrome complicated by Castleman disease.
- Rituximab (anti-CD20 antibody)
- Thrombopoietin receptor agonists romiplostim and eltrombopag: for patients with persistent thrombocytopenia.
・Glucocorticoids are the first line treatment for patients with TAFRO syndrome, with CsA recommended as the second line treatment for patients refractory to glucocorticoids.
・If CsA is contraindicated, as in patients with renal insufficiency, tocilizumab or rituximab is recommended.
・Plasma exchange, cyclophosphamide, combination chemotherapy such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), thalidomide and lenalidomide, have been successful in the treatment of selected patients.
・Splenectomy and high-dose gamma-globulin have not been shown effective.